
Overview
Quantum chemistry is currently at the exciting stage where one can make accurate predictions about many molecular properties. At the same time, one constantly encounters chemical phenomena that are still poorly understood and poorly described by existing theoretical techniques. The Van Voorhis group develops new methods that make reliable predictions about real systems for which existing techniques are inadequate. Several of our active research topics are summarized below.
First Principles Description of Electron Transfer 
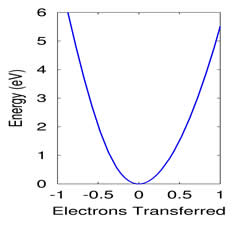
Electron transfer (ET) reactions are perhaps the most basic chemical reactions. However, it is difficult to model the changes in electronic structure that accompany ET because the reaction pushes the electrons out of equilibrium, and most programs are specifically designed for the equilibrium case. We have developed a scheme that allows us to directly solve for non-equilibrium states with partial or total ET within the context of constrained density functional theory (DFT). This allows us to probe the energy profile encountered by the reactive electron. For example, the graph at right displays the energy required to transfer electrons onto or off of the amine group in dimethylamino-benzonitrile (DMABN). The asymmetry results from the fact that the amine group is a better electron donor than acceptor. We are in the process of applying this method to a number of problems. Our initial results indicate that this constrained DFT formalism is essentially complementary to time-dependent DFT (TDDFT). TDDFT is well-suited to short-range ET, but fails at long range; constrained DFT works beautifully for long range ET, but is poorly defined at short distances. We are particularly interested in using these methods to parameterize model Hamiltonians for cases where DFT fails.
Modeling Real-Time Electron Dynamics
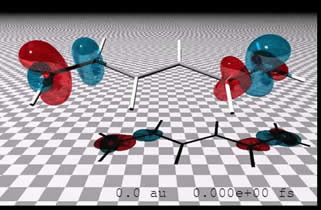
Ab initio methods tend to focus the lion's share of attention on the description of electronic structure. However, there are a variety of systems where a focus on the electron dynamics is extremely fruitful. This is true, for example, in conducting organic polymers and crystals - where it is charge migration that leads conductivity - and in photosynthetic and photovoltaic systems - where excited state energy transfer determines the efficiency. We have developed an efficient means of performing real time TDDFT simulations. This allows us to study the ultrafast electron dynamics that lead to charge transfer. For example, the movie at left illustrates ET in hexatriene. Red indicates charge accumulation, and blue shows charge depletion. The initial state arises from transferring a single charge from one end of the "molecular wire" to the other. The movie shows the first few femtoseconds of relaxation. By extending this to longer times and larger "wires" we are able to approximate the steady-state dynamics probed in current-voltage experiments. We are in the process of developing a theory that directly accesses these steady states. We are also studying the real time dynamics of resonant energy transfer between chromophores using TDDFT.
Nuclear Dynamics Triggered by Electron Motion
For the first several femtoseconds, it is reasonable to deal with the electron dynamics by themselves - the nuclei simply don't have time to respond! However, at long times, vibrational motion plays a key role in the ET process: phonons provide a means of dissipating excess energy, and conformational changes of the molecule can either promote or hinder the ET. We hope to use the electron dynamics at relatively short times to extract information about these key vibrational processes. On the one hand, we are developing semiclassical techniques that are capable of dealing with non-Born-Oppenheimer effects, such as the branching of the nuclear trajectory based on the fate of the electronic degrees of freedom. This is typically accounted for by a surface-hopping approximation, but our semiclassical formalism includes this in a natural way. We are also keenly interested in developing tools that will allow us to glean information about low-frequency, large amplitude motions from data collected at short times. This is crucial for coupled electron-nuclear systems because of the disparity of time scales for electronic and vibrational motion.
Electron Spins and Molecular Magnetism
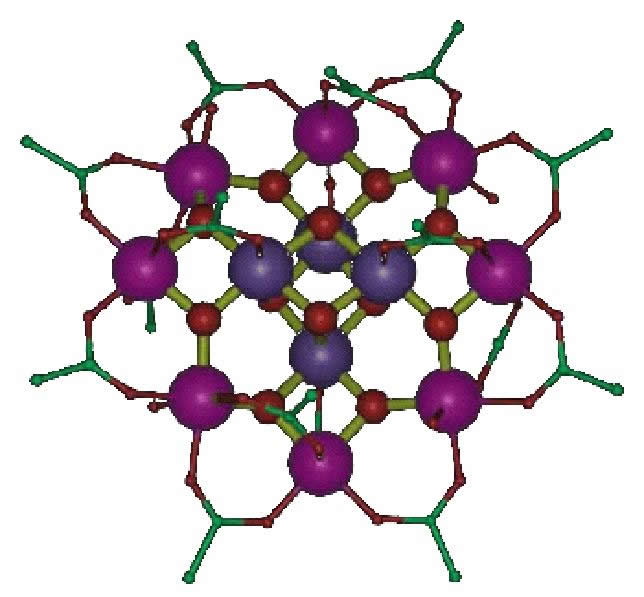
Molecular magnetism is currently an exciting area in chemical physics because of the technological promise of single molecule magnets, like the Mn12 molecule shown at right. This cluster has a ground state spin of S=10, which generates a large magnetic moment that could (in principle) be used to store data. Theoretical predictions on a molecule of this type are exceedingly difficult due to the size of the system and the delicate correlations between the different electron spins within a cluster. We have developed a simple method for predicting the anisotropy (i.e. the energy barrier between "spins up" and "spins down") of molecules like this that utilizes only information gleaned from single-ion fragments. This gives a good rule of thumb for anticipating how different building blocks can be assembled to yield a large barrier. We are also exploring ways of calculating Heisenberg exchange parameters in these clusters using the constrained DFT methodology described above. These investigations are part of the greater goal of understanding the role of electron spin in molecular bonding and electronic structure.